

Welcome to the working group
Brassicaceae polyploidy
The main topics addressed by our team
are microevolutionary and speciation processes in diploid-polyploid complexes belonging to the Brassicaceae family. Speciation both at the diploid and polyploid levels is examined, elucidating the role of genome merge, duplication, and hybridisation, together with geographical and ecological separation. The questions addressed have broader relevance, beyond the studied genera, and the answers can help us better understand processes underlying polyploid speciation as such. Methods applied to address such questions include both conventional molecular systematics markers, as well as methods based on the Next Generation Sequencing (NGS), namely restriction site associated DNA sequencing (RAD-seq) and combination of target enrichment of low-copy nuclear genes and genome skimming (Hyb-Seq). Using the multi-marker approach we expect to increase the robustness of the results. As a complement, flow-cytometric, morphometric, ecological and climatic niche analyses are employed. In order to achieve these goals, we have chosen four genera from three different tribes of the Brassicaceae family (Alysseae [Alyssum, Odontarrhena], Camelineae [Arabidopsis], Cardamineae [Cardamine]), representing different evolutionary lineages in this family.
News… in progress
- Paper about the climatic niche evolution in relation to the lineages published
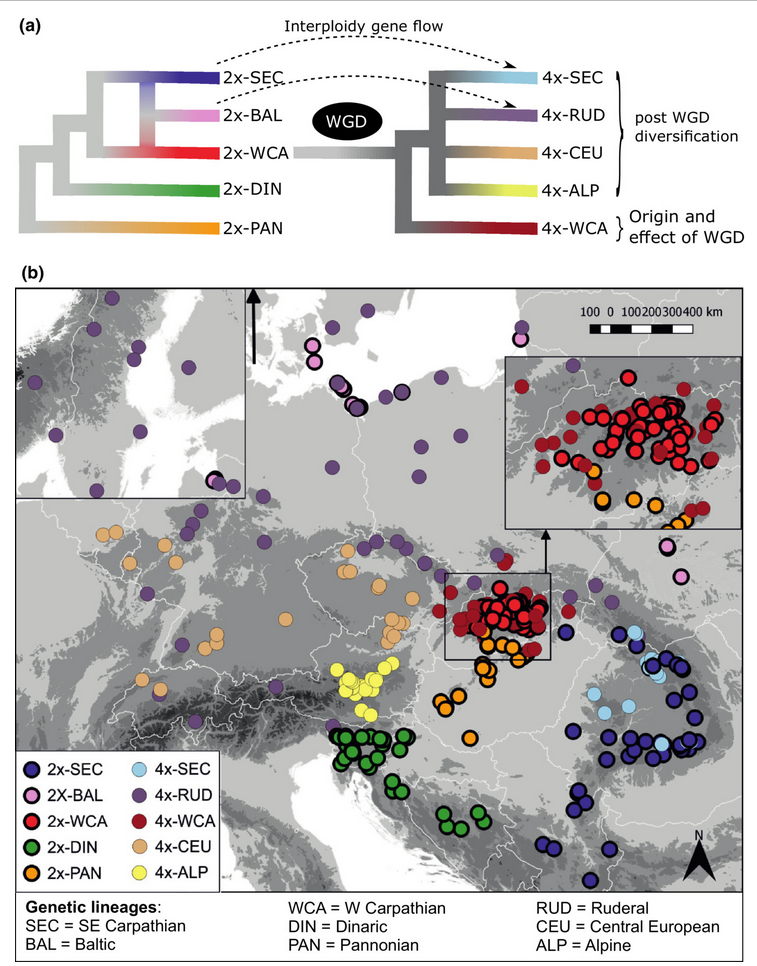 Our study about the importance of considering the evolutionary history of polyploids when assessing climatic […]
Our study about the importance of considering the evolutionary history of polyploids when assessing climatic […] - PhD theses about the evolution of Arabidopsis in Europe finally defended!
 After a couple of years of collecting data and publishing the results, the PhD thesis […]
After a couple of years of collecting data and publishing the results, the PhD thesis […]